Artículo
Crescentic and necrotising glomerulonephritis: A rare histological manifestation of Alport syndrome

Fecha de publicación:
07/2015
Editorial:
B M J Publishing Group
Revista:
Journal Of Clinical Pathology
ISSN:
0021-9746
Idioma:
Inglés
Tipo de recurso:
Artículo publicado
Clasificación temática:
Resumen
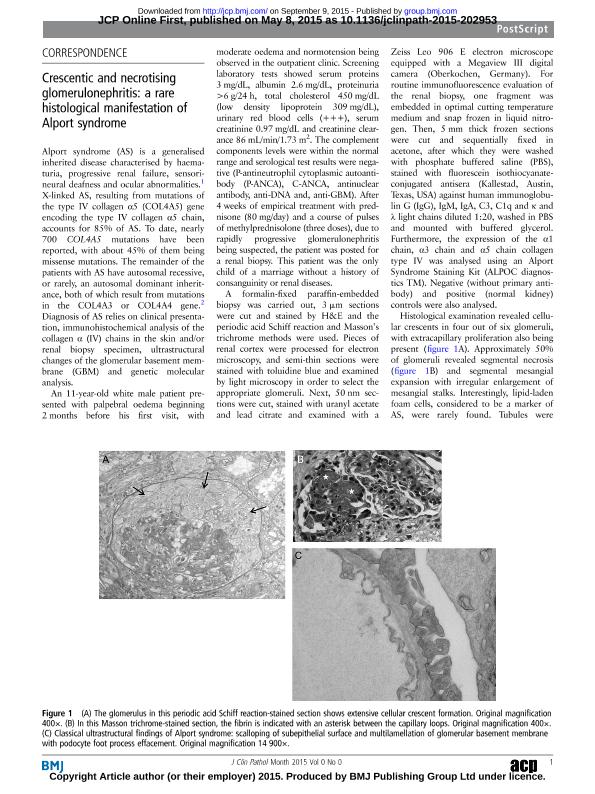
Alport Syndrome (AS) is a generalized inherited disease characterized by hematuria, progressive renal failure, sensorineural deafness and ocular abnormalities [1]. X-linked AS, resulting from mutations of the type IV collagen alpha 5 (COL4A5) gene encoding the type IV collagen α5 chain, accounts for 85% of AS. To dates, nearly 700 COL4A5 mutations have been reported, with about 45% of COL4A5 mutations being missense mutations. The remainder of the AS patients have autosomal recessive, or rarely, an autosomal dominant inheritance, both of which result from mutations in the COL4A3 or COL4A4 gene [2]. Diagnosis of AS relies on clinical presentation, immunohistochemical analysis of the collagen α (IV) chains in the skin and/or renal biopsy specimen, ultrastructural changes of the glomerular basement membrane (GBM) and genetic molecular analysis.An 11-year-old white male presented with palpebral edema beginning two months before his first visit, with moderate edema and normotension being observed in the outpatient clinic. Screening laboratory tests showed serum proteins 3 mg/dl, albumin 2.6 mg/dl, proteinuria >6 gr/24 h, total cholesterol 450 mg/dl (LDL 309 mg/dl), urinary red blood cells (+++), serum creatinine 0.97 mg/dl and creatinine clearance 86 ml/min/1.73m2. The complement components levels were within the normal range, and serological test results were negative (P-ANCA, C-ANCA, ANA, anti-DNA and, anti-GBM). After four weeks of empirical treatment with prednisone (80 mg/d) and a course of pulses of methylprednisolone (3 doses), due to rapidly progressive glomerulonephritis being suspected, the patient was derived for a renal biopsy. This patient was the only child of a marriage without a history of consanguinity or renal diseases. A formalin-fixed paraffin-embedded biopsy was carried out and, 3 um sections were cut and stained by hematoxylin and eosin and, the PAS and Masson´s trichrome methods were used. Pieces of renal cortex were processed for electron microscopy and, semithin sections were stained with toluidine blue and examined by light microscopy in order to select appropriate glomeruli. Next, 50 nm sections were cut, stained with uranyl acetate and lead citrate, and examined with a Zeiss Leo 906 E electron microscope equipped with a Megaview III digital camera (Oberkochen, Germany). For routine immunofluorescence evaluation of the renal biopsy, one fragment was embedded in OCT médium and, snap frozen in liquid nitrogen. Then, 5 mm thick frozen sections were cut and sequentially fixed in acetone, after which they were washed with PBS, stained with FITC-conjugated antisera (Kallestad, Austin, TX, USA) against human IgG, IgM, IgA, C3, C1q and κ and λ light chains diluted 1:20, washed in PBS, and mounted with buffered glycerol. Futhermore, the expression of the α1 chain, α3 chain and α5 chain collagen type IV was analyzed using an Alport Syndrome Staining Kit (ALPOC diagnostics TM). Negative (without primary antibody) and positive (normal kidney) controls were also carried out. A histological examination revealed cellular crescents in four out of six glomeruli, with extracapillary proliferation also present (Figure 1A). Approximately 50 % of glomeruli revealed segmental necrosis (Figure 1B) and segmental mesangial expansion with irregular enlargement of mesangial stalks. Interestingly, lipid-laden foam cells, considered to be a marker of AS, were rarely found. Tubules were normal, except for the presence of occasional red blood casts, and no significant vascular changes were noted.Ultrastructural examination of the glomerular capillary loops showed a diffuse and abnormal architectural organization of the GBMs, characterized by frequent electron-lucent areas with a frequently lamellated and ?basket-weave? appearance in glomeruli with or without crescents (Figure 1C). No immune-type electron-dense deposits were identified in different renal compartments but there was an extensive effacement of podocyte processes. The immunofluorescence studies for IgG, IgA, IgM, C3 and C1q and κ and λ light chains revealed no significant deposition of these reactants in the glomeruli, tubular basement membranes or the interstitium. Results of the α3 or α5 collagen IV immunostaining were negative, contrasting with the α1(IV) labeling of GBM and tubular basement membranes (Figure 2 A-B-C). Hypoacusia was confirmed by audiometry and DNA analysis using whole exome sequencing confirmed that the proband carried the c.G3508A mutation in COL4A5, which is considered to be pathogenic and is known to cause X-linked AS (Figure 2D). Oral cyclophosphamide was administered for 8 weeks and seven months after renal biopsy, the patient had SCr 1.34 mg/dl, and proteinuria 324 mg/24 hrs, so dual antiproteinuric therapy (ACEI/ARB), being added. This case illustrated a rare histological manifestation of AS, which is a hereditary disease of GBM deriving from a defect in the gene encoding for type IV collagen α-chain isoforms and, clinically characterized by a progressive nephropathy often associated with sensorineural deafness and ocular abnormalities [1,2]. Among the different light microscopy patterns shown in AS, necrotizing and crescentic glomerulonephritis are rarely found in this syndrome with only a few cases reported to date [1, 2, 3, 4, 5, 6]. However, this pattern of glomerulonephritis is well known in transplant kidneys [7]. The presence found of glomerular crescents and fibrinoid necrosis raises the possibility of the other causes being responsible for the crescentic glomerulonephritis and a superimposed ANCA associated or pauci-immune type of crescentic glomerulonephritis can not be ruled out, especially if we consider that approximately 20% of the pauci-immune type of crescentic glomerulonephritis may be associated with negative ANCA titters[8, 9,10]. However, the ultrastructural analysis showed characteristic of AS, as confirmed by immunodetection of collagen type IV chains. Finally the mutation was pathogenic of X-linked AS as expected [11].In summary, a careful search for CsGN, particularly in paediatric populations is justified, because a nephrotic syndrome in the childhood does not rule out a CsGN. In this context the possibility of AS should be considered. Consequently, ultrastructural analysis represents an indispensable tool for a definitive nosological diagnosis.
Palabras clave:
DIAGNOSTICS
,
ELECTRON MICROSCOPY
,
KIDNEY
Archivos asociados
Tamaño:
256.0Kb
Formato:
PDF
Licencia
 Excepto donde se diga explícitamente, este item se publica bajo la siguiente descripción:
Creative Commons Attribution-NonCommercial-ShareAlike 2.5 Unported (CC BY-NC-SA 2.5)
Excepto donde se diga explícitamente, este item se publica bajo la siguiente descripción:
Creative Commons Attribution-NonCommercial-ShareAlike 2.5 Unported (CC BY-NC-SA 2.5)
Identificadores
Colecciones
Articulos(INICSA)
Articulos de INSTITUTO DE INVESTIGACIONES EN CIENCIAS DE LA SALUD
Articulos de INSTITUTO DE INVESTIGACIONES EN CIENCIAS DE LA SALUD
Citación
Gabriela Deisi, Moyano Crespo; Torres, Alicia Ines; Mukdsi, Jorge Humberto; Crescentic and necrotising glomerulonephritis: A rare histological manifestation of Alport syndrome; B M J Publishing Group; Journal Of Clinical Pathology; 68; 7; 7-2015; 581-583
Compartir


Altmétricas